MTM¶
About¶
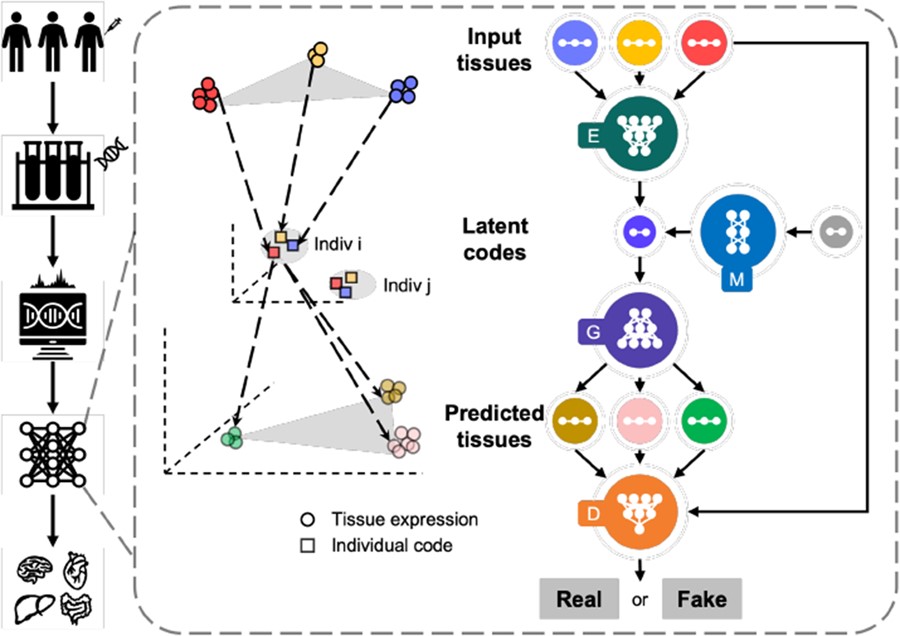
MTM (Multi-tissue Transcriptome Mapping) is a unified deep multi-task learning framework that predicts tissue-specific gene expression profiles using any available tissue expression profile from the same donor, such as blood gene expression.
Installation¶
Software Requirements¶
Python 3.8
PyTorch 1.10.2
NumPy 1.20.3
Pandas 1.2.4
scikit-learn 0.24.2
Obtain MTM¶
Clone the repository:
git clone https://github.com/yangence/MTM.git
Enter the project directory:
cd MTM
Usage¶
Input Files¶
Download the following data from the GTEx Portal:
Expression data:
GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_tpm.gct.gzSample attributes:
GTEx_Analysis_v8_Annotations_SampleAttributesDS.txt
Data Filtering¶
Filter the downloaded data with the following criteria:
Tissue types with at least 50 samples
Individuals with at least 2 tissue samples
Genes of interest (for example, protein-coding genes)
Add the donor ID to the sample attributes file as the Subject_id column.
Expected files for training:
File |
Description |
|---|---|
|
Expression matrix (rows = samples, columns = genes) |
|
Sample attributes with tissue type ( |
|
Filtered gene identifiers |
|
Filtered individual identifiers |
|
Filtered tissue types |
Train¶
python train.py \
--input_dir ../input_dir \
--expr GTEx_expr.txt \
--sample_attr GTEx_sample_attributes.txt \
--gene_id GTEx_gene_id.txt \
--indiv_id GTEx_individual_id.txt \
--tissue_type GTEx_tissue_type.txt \
--device "cuda:0" \
--output_dir ../output_dir
Main outputs:
Trained model checkpoint under
../${output_dir}/models/Training and validation splits under
../${output_dir}/data_split/
Predict¶
python predict.py \
--expr GTEx_expr.txt \
--sample_attr GTEx_sample_attributes.txt \
--gene_id GTEx_gene_id.txt \
--indiv_id GTEx_individual_id.txt \
--tissue_type GTEx_tissue_type.txt \
--input_expr GTEx_expr.val_set.Whole_Blood.txt \
--input_tissue_type "Whole_Blood" \
--output_tissue_type "Lung" \
--model_path ../output_dir/models/model_ckpt.tar \
--output_expr ../output_dir/predicted/GTEx_expr.val_set.Whole_Blood.to.Lung.txt
Main output:
Predicted expression profile file for the target tissue
Example Workflow¶
Preprocess GTEx data by filtering tissues, individuals, and genes.
Train MTM with
train.py.Prepare source-tissue input expression (for example
Whole_Blood) for validation individuals.Run prediction with
predict.pyfor the target tissue.
License¶
MTM is licensed under the terms included in its repository LICENSE file:
Citation¶
If you use MTM in your research, cite:
He G, Chen M, Bian Y, et al. MTM: a multi-task learning framework to predict individualized tissue gene expression profiles. Bioinformatics, 2023, 39(6): btad363.